Notion d'espèce au travers du séquençage d'ADN



Notion d'espèce au travers du séquençage d'ADN
I. Rappel sur le vocabulaire de la génomique
Un gène est une portion d’ADN occupant un locus sur un chromosome, fait d’une succession de nucléotides ordonnés et c’est cet ordre qui est ensuite séquencé, transcrit en un ARN pré-messager qui sera traduit en protéine. Ce gène ne va être transcrit que s’il dispose d’une séquence promotrice qui sera activée et il peut être surexprimé via des facteurs cis et trans ou au contraire s’éteindre.
Ce gène peut présenter plusieurs versions ou allèles qui définissent la diversité au sein d’une espèce. Au sein d’une espèce, les génotypes varient : nous n’avons pas tous les mêmes allèles d’où des phénotypes variants. La biodiversité qu’on observe au sein d’une espèce s’explique par des phénotypes gouvernés par le génotype mais aussi part l’environnement.
Ces gènes forment un génome. Dans l’espèce humaine, sur nos 46 chromosomes, on a pu séquencer la totalité de l’ADN, ce qui représente 3,2 milliards de paires de bases. Dans ces bases, après séquençage, on a constaté qu’il y avait 23 000 gènes, ce qui est très peu au regard de la quantité : 2 % de notre ADN. Ces 23 000 gènes permettent de coder des protéines : un gène peut coder plusieurs protéines grâce à une maturation différentielle de l’ARN pré-messager en ARN messager. On considère que notre protéome, la totalité de nos protéines, compte à peu près 300 000 protéines.
II. Histoire du séquençage
Comment notre génome humain a pu être séquencé ? Séquencer l’ADN c’est comprendre l’ordre des nucléotides sur un fragment d’ADN, puis à grande échelle sur les 3,2 giga paires de bases qu’on possède.
A. La méthode de Maxam et Gilbert
Dans les années 1970, Maxam et Gilbert ont l’idée d’utiliser un traceur pour séquencer les gènes : le 32P, un radioisotope. Ils prennent une séquence simple brin d’ADN et l’amplifient. Pour cela, on peut employer la méthode PCR, une réaction de polymérisation en chaîne qui permet d’avoir le morceau d’ADN voulu en beaucoup d’exemplaires. Maxam et Gilbert partent avec plusieurs fois le même morceau d’ADN, pris en simple brin, et y accrochent un 32P. Ensuite, ils répartissent leurs clones de simples brins dans 4 tubes à essai et vont, dans chacun des tubes, détruire de manière spécifique des bases.
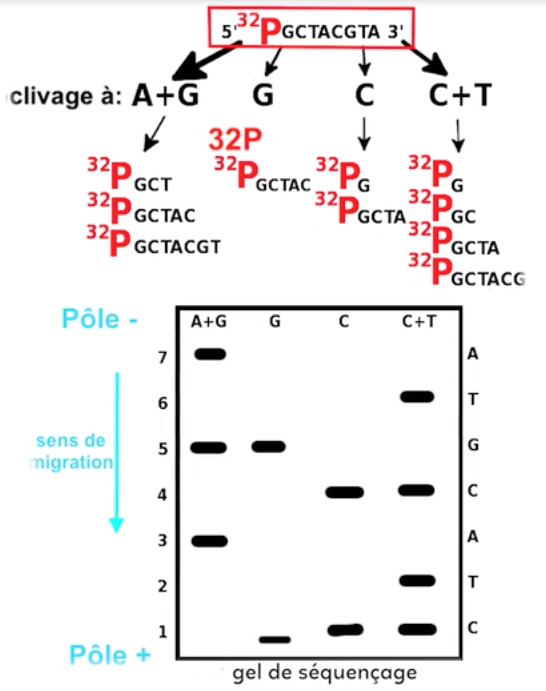
Sur ce schéma, on part avec un ADN simple brin radiomarqué avec le 32P en extrémité 5’ et il est séquencé (initialement on ne connaît pas la séquence, on veut la connaître). On met cette séquence qu’on ignore dans 4 tubes à essai :
– Le 1er contient des produits qui dénaturent spécifiquement les bases A (adénine) et G (guanine).
– Le 2e contient un produit dénaturant la guanine exclusivement.
– Le 3e contient un produit qui dénature la cytosine exclusivement.
– Le 4e contient un produit qui dénature la cytosine et la thymine.
On laisse la dénaturation se faire et ensuite on récupère les différents fragments.
Par exemple, on voit que dans le tube contenant un produit qui dénature A et G, si on regarde la séquence de départ, elle va couper juste après G, juste après A, puis de nouveau G, puis de nouveau A : on obtient des fragments de plusieurs tailles.
On dépose ces derniers dans un gel de polyacrylamide dans lequel on a fait des petits puits, chaque contenu de tube à essai est déposé dans un puits. On met ensuite en place un générateur qui crée un courant électrique : les fragments chargés négativement vont migrer vers le pôle +. Plus le fragment est petit plus il migre loin. Si on regarde le résultat du gel on constate que le fragment qui a migré le plus loin provient du tube qui contenait un produit dénaturant G. Cela signifie que le premier nucléotide de la séquence est le nucléotide G. Ensuite, on constate qu’on a deux petits fragments provenant des tubes qui contenaient des produits dénaturant C et C+T. On en déduit que le deuxième nucléotide est le nucléotide C. On poursuit dans cette logique la lecture du résultat du gel : vient ensuite dans la séquence le nucléotide T, puis le nucléotide A, ainsi de suite.
De cette manière Maxam et Gilbert on pu séquencer de courts fragments d’ADN.
B. La méthode de Sanger
C’est une autre méthode qui date des années 1970. À l’aide d’une ADN polymérase, l’idée est de synthétiser progressivement le brin d’ADN à séquencer. Pour cela Sanger utilise des ddNTPs (di-désoxyribonucléotides). Un ddNTP a ses deux fonctions OH en 2’ et 3’ qui ont été enlevées de sorte qu’il ne peut plus participer à uneéelongation de l’ADN. Ces ddNTP sont marqués avec un fluorochrome. Le ddNTP, lorsque c’est de l’adénine par exemple, va achever la polymérisation de l’ADN lorsqu’il est incorporé. Pour réaliser cette méthode, il faut donc une ADN polymérase, une amorce, et pour continuer de polymériser, il faut des dNTP.
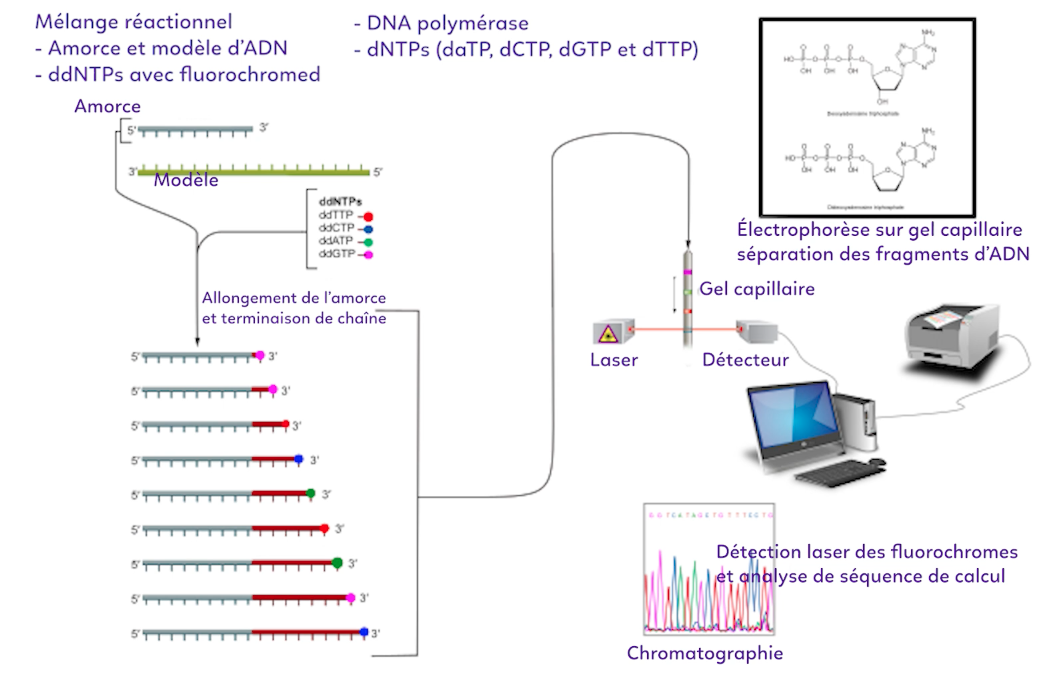
La méthode de Sanger consiste à récupérer des ddNTP marqués par 4 fluorochromes avec une couleur par base azotée : pour le ddTTP (thymine), pour le ddCTP (cytosine), pour le ddGTP (guanine), pour le ddATP (adénine).
Sanger prend une amorce, ainsi que la séquence qu’il souhaite connaître, en n’ayant pris qu’un seul des deux brins, et ajoute dans un tube à essai les ddNTPs marqués avec des fluorochromes ainsi que des dNTP. Il lance ensuite la polymérisation.
Il va obtenir plusieurs fragments d’ADN de longueurs différentes, chacune se terminant par un fluorochrome donné. Le fragment le plus court permet de connaître le premier nucléotide et ainsi de suite jusqu’au fragment le plus long : cela permet de reconstituer la séquence en entier.
Cette technique de séquençage est aussi performante, comme celle de Sanger et Gilbert, mais dans les années 1990, il fallait environ 5 jours pour séquencer 800 nucléotides.
C. Des années 2000 à nos jours
Dans les années 2000, apparaissent des techniques plus performantes mais assez complexes, dites de shotgun. On a beaucoup utilisé la PCR pour amplifier des séquences au sein de vecteurs qui peuvent être des bactéries ou des levures (pour des fragments plus longs). Il y alors eu un consortium de plusieurs laboratoires, HGP (Human Genome Project), qui se sont lancés dans la course au séquençage du génome humain. Puis une entreprise privée, Celera Genomics, tenue par Craig Venter a été fondée en 1998 dans le but de générer puis commercialiser des informations génétiques. Leurs recherches furent parmi les premières à montrer la faisabilité de la technique de séquençage par shotgun. On est ainsi parvenu à établir la totalité de l’ordre des nucléotides du génome humain, c’est-à-dire à le séquencer presque entièrement (sauf les zones à haute répétabilité).
Actuellement, on utilise des méthodes encore plus performantes, comme le séquençage à haut débit : on peut séquencer plusieurs milliards de paires de base en une journée. On peut aussi utiliser des nanopores (on fait passer l’ADN simple brin et on va pouvoir séquencer, « découper », en fonction de champ électrique et déterminer la séquence).
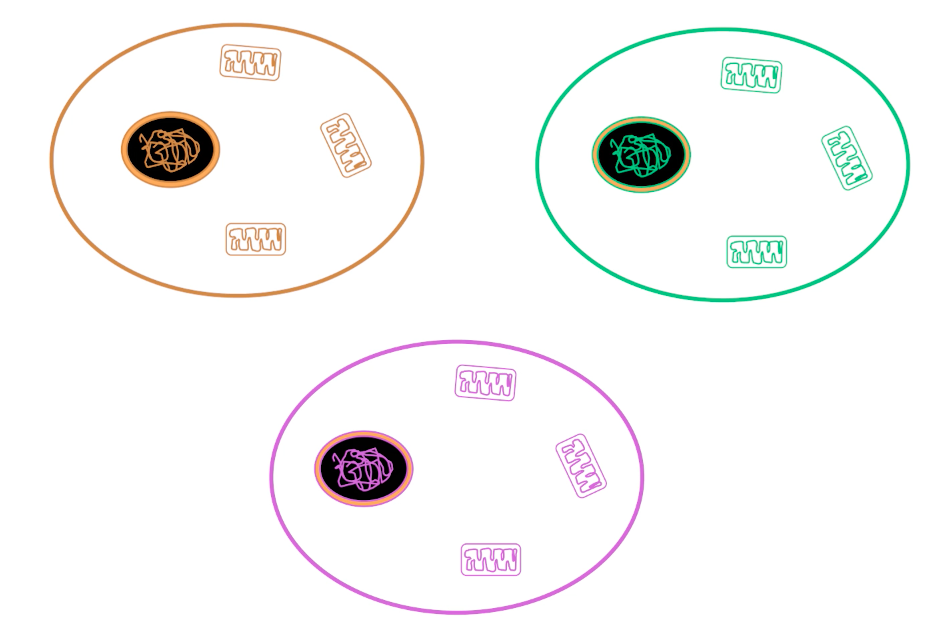
Pour terminer, on cite le barcoding. Dans ce schéma, on voit trois organismes unicellulaires. On a trouvé, notamment dans le génome des organites, que la CO1, la première unité de la cytochrome oxydase présentait au sein d’une espèce une très faible variabilité (seulement 3 %). Le barcode a pour principe de séquencer juste cette petite séquence d’ADN présent dans les mitochondries des trois individus dont on cherche à savoir s’ils sont ou non de la même espèce. Si on trouve moins de 3 % de différence, on pourra conclure que ces trois individus appartiennent à la même espèce.
Conclusion
Le séquençage ADN a été très long à se mettre en place depuis les années 1970. En 40 ans, il y a eu une progression très importante des méthodes. Celles-ci sont utilisées aujourd’hui notamment pour analyser des échantillons d’ADN provenant de l’environnement (exemples : sol, intestins, flaque d’eau, etc.) afin d’étudier la biodiversité microbienne de ces environnements. C’est ce qu’on appelle la métagénomique et ainsi séquencer l’ADN permet de comprendre le vivant et la diversité des espèces.